
Middle author bioinformatics
your friendly neighborhood bioinformaticians
CORE INFRASTRUCTURE & OPERATIONAL CAPABILITIES
| Dedicated Compute Infrastructure | Our private analytical server (nicknamed The Belly) provides high-memory, high-storage local compute capacity independent of institutional queues or shared cluster contention. This enables rapid turnaround, controlled environments, and reliable execution of long-running genomics analyses. |
| Scalable Cloud Computing | We also rely on Amazon Web Services (AWS), which enables elastic compute for large-scale genomics, secure long-term storage, and deployment of client-facing dashboards and web applications. Projects can scale seamlessly from pilot datasets to production-level studies. |
| Secure Data Management & Transfer | Structured intake, storage, backup, and controlled sharing of client data. Versioned outputs, reproducible organization, and secure cloud-based delivery ensure integrity, traceability, and professional stewardship of sensitive research materials. |
| Reproducible Workflow Environments | Containerized and environment-controlled analytical pipelines with fully documented software versions and parameters. Analyses remain auditable, rerunnable, and publication-ready months to years after project completion. |
| Transparent Project Management & Communication | Structured coordination through Basecamp, including milestones, deliverables, file exchange, and discussion tracking. This provides clear timelines, progress visibility, and reduced uncertainty for collaborators. |
| Custom Software & Web Applications | Development of tailored dashboards, visualization systems, and secure analysis portals that allow interactive exploration of genomic and metagenomic results through modern cloud infrastructure. |
| Scientific Expertise & Confidential Interpretation | Direct collaboration with PhD-level bioinformaticians providing guidance on experimental design, analytical strategy, and biological interpretation. All projects are conducted under formal confidentiality protections, including non-disclosure agreements (NDAs) and data-handling agreements, ensuring complete privacy of client data, results, and intellectual property. |
| Long-Term Data Stewardship | Archival preservation of project data and analytical outputs, enabling future reanalysis, follow-up studies, and continuity beyond the duration of a single grant or trainee appointment. |
Excellence
Benevolence
Strategic Partners








Services
1. What's-in-the-bag? 🔎🧫
Strain-typing and serotyping
Multi-Locus sequence typing (MLST)
Prokaryote vs eukaryote identification
2. Resistance-is-futile! ✊
Antimicrobial resistance discovery
Variant-calling and mutation prediction
Annotation of biosynthetic gene clusters
3. Get-me-the-microbes! 🪆
Bacterial and Archaeal Genome Reconstruction from Mixed Samples
Viral and phage genome discovery
Genome evaluation and annotation
4. Dude-where's-my-transposon? 🛫✂️🛬
Analysis of Tn-Seq datasets
Identification of insertions and genes affected
Functional annotation of impacted genes
5. Everything-RNA 🧬
Differential Expression Analysis
Transcriptome Assembly
Identification and Removal of Ribosomal RNA (rRNA)
6. Amplicon-and-microbiome 🧮
Generation of Amplicon Sequence Variants (ASVs) or Operational Taxonomic Units (OTUs)
16S rRNA and Internal Transcribed Spacer Sequencing
Alpha and Beta Diversity Metrics
7. Genome-assembly 🪢
Prokaryotic and eukaryotic Genomes
de novo annotation of prokaryotic genomes
Transcriptome and proteome-guided annotation of eukaryotic genomes
8. Structural-biology 🏗️
de novo protein folding and structural prediction
Domain classification and functional annotation
Identification of structural homologs

Selected Publications and Software contributions



Garber, A. I., Armbruster, C. R., Lee, S. E., Cooper, V. S., Bomberger, J. M., & McAllister, S. M. (2022). SprayNPray: user-friendly taxonomic profiling of genome and metagenome contigs. _BMC Genomics_, 23(1), 202.
link to article
link to software.



Syberg-Olsen, M. J., Garber, A. I., Keeling, P. J., McCutcheon, J. P., & Husnik, F. (2022). Pseudofinder: detection of pseudogenes in prokaryotic genomes. _Molecular Biology and Evolution_.
link to article.
link to software.



Ramírez, G. A., Keshri, J., Vahrson, I., Garber, A. I., Berrang, M. E., Cox, N. A., González-Cerón, F., Aggrey, S. E., & Oakley, B. B. (2022). Cecal Microbial Hydrogen Cycling Potential Is Linked to Feed Efficiency Phenotypes in Chickens. _Frontiers in Veterinary Science_, 9, 904698.
link to article




• Garber, A. I., Kupper, M., Laetsch, D. R., Weldon, S. R., Ladinsky, M. S., Bjorkman, P. J., & McCutcheon, J. P. (2021). The Evolution of Interdependence in a Four-Way Mealybug Symbiosis. _Genome Biology and Evolution_, 13(8).
link to article



Garber, A. I., Zehnpfennig, J. R., Sheik, C. S., Henson, M. W., Ramírez, G. A., Mahon, A. R., Halanych, K. M., & Learman, D. R. (2021). Metagenomics of Antarctic Marine Sediment Reveals Potential for Diverse Chemolithoautotrophy. _mSphere_, 6(6), e0077021.
link to article
link to software.



• Garber A. I., Nealson KH, Okamoto A, McAllister SM, Chan CS, Barco RA, Merino N (2020) FeGenie: a comprehensive tool for the identification of iron genes and iron gene neighborhoods in genome and metagenome assemblies. _Frontiers in Microbiology_ 11:37.
link to article
link to software.



• Ramírez GA, Garber A. I., Lecoeuvre A, D’Angelo T, Wheat CG, Orcutt BN (2019) Ecology of Subseafloor Crustal Biofilms. _Frontiers in Microbiology_.
link to article.
link to software


• Armbruster, C. R., Marshall, C. W., Garber, A. I., Melvin, J. A., Zemke, A. C., Moore, J., Zamora, P. F., Li, K., Fritz, I. L., Manko, C. D., Weaver, M. L., Gaston, J. R., Morris, A., Methé, B., DePas, W. H., Lee, S. E., Cooper, V. S., & Bomberger, J. M. (2021). Adaptation and genomic erosion in fragmented Pseudomonas aeruginosa populations in the sinuses of people with cystic fibrosis. Cell Reports, 37(3), 109829.
link to article



• Keffer, J. L., McAllister, S. M., Garber, A. I., Hallahan, B. J., Sutherland, M. C., Rozovsky, S., & Chan, C. S. (2021). Iron Oxidation by a Fused Cytochrome-Porin Common to Diverse Iron-Oxidizing Bacteria. _mBio_, 12(4), e0107421.
link to article
• BagOfTricks: A set of short-to-medium length software tools for various bioinformatics tasks. This is a growing list of tools that I make for various projects, which I then make broadly available to others for use.
link to software.
Please reach out to [email protected] for
information on how to securely transfer data to the MAB server.
Data drives can also be mailed directly to the following address:
Middle Author Bioinformatics
1326 E Avalon Dr.
Phoenix, AZ 85014
United States of AmericaPhone Number: (818) 324-1145
Leadership

Arkadiy Garber: Co-Founder and Principal Bioinformatician
I founded this company to provide bioinformatics seamlessly to research labs across the world. We can provide a wide range of analyses to meet the flexible timelines, budgets, and needs of academic and industrial labs.

Dr. Vaughn Cooper: Co-Founder, Advisor
Vaughn Cooper is an evolutionary microbiologist and Professor at the University of Pittsburgh. He is co-Founder and Scientific Advisor of both SeqCoast Genomics and Middle Author Bioinformatics, which work together to provide advanced genome-scale sequencing and analyses using intuitive and accessible processes. Vaughn believes that there has never been a better time to be a microbiologist or geneticist thanks to unprecedented advances in sequencing and computational technology, and he is committed to democratizing access to these powerful tools.

Dr. Jean-Paul Baquiran: Director of Operations
Jean-Paul (JP) is Chief Scientific Officer of Biologic Environmental, a firm specializing in sustainable waste management. Jean-Paul has a long career that reflects his passion in bioremediation and sustainability. His background in microbial and molecular biology, in combination with strong business and leadership skills, makes JP an important asset to this company.

Dr. Gustavo Ramírez: Senior ScientistWhen he is not scouring the depths of the ocean for enigmatic micro and macrofauna, Gustavo Ramírez runs a lab at CSULA, which focuses on microbial ecology of the marine subsurface and host-microbe interactions in human-built environments. As our lead consultant for environmental sciences and microbial ecology, Gus leverages his expertise in the natural sciences with a strong background in data science, data visualization, and biostatistical analyses.

Dr. Michael Pavia: Senior BioinformaticianMike has nearly a decade of experience in microbiology. Much of this decade has been spent at the command line, wrangling hundreds of metagenomes and metatranscriptomes. A true believer is scientific dissemination and improving access to science for all, Mike founded a podcast, Mikroscope, designed to deliver cutting-edge scientific discoveries to the general public. He currently works with virologists at ASU to leverage machine-learning in epidemiology.

Faiza Rafi: Junior BioinformaticianFaiza earned her bachelor’s degree in Biotechnology from the City College of New York, where early experimental research in bacterial systems shaped her interest in molecular biology and genomics. Exposure to computational biology at the New York Genome Center led her to pursue a master’s degree in Bioinformatics at NYU Tandon. She is passionate about applying computational approaches to investigate living systems, with interests spanning microbial genomics, adaptation, and molecular mechanisms.

Rebecca Reinking-Herd: Assistant Project ManagerBecca earned her bachelor’s degrees in Biology and Physics from Carleton College, and began her career by working for Teach for America in Pueblo, Colorado. After six years of teaching and administration, she returned to science for a master’s degree in Biology, specializing in Immunology. With her backgrounds in both science and education, Becca is passionate about developing an array of scientific tools to enhance comprehensive scientific discovery as well as promoting scientific communication for broader public understanding.

Dr. Kara Schmidlin: Bioinformatician
Specializing in bioinformatics, evolutionary genetics, and barcoded evolutionDr. Kara Schmidlin is a bioinformatician and evolutionary geneticist whose work focuses on understanding how organisms adapt to environmental and drug-induced stress. Her research combines high-throughput sequencing, barcoded evolution experiments, and quantitative fitness modeling to study drug resistance, collateral sensitivity, and genotype–phenotype–fitness relationships. She brings deep expertise in experimental design, data analysis, and interpretation of complex evolutionary datasets..

Arkadiy Garber: Lead Bioinformatician
Specializing in microbial symbioses, geomicrobiology, and biochemistryArkadiy enjoys programming and designing/implementing bioinformatics pipelines and software packages. His research spans geomicrobiology, microbial ecology, environmental and clinical microbiology, and evolutionary biology. As humanity generates more and more biological sequence data, we increase potential to make novel discoveries that improve our understanding of fundamental biology and help to implement biotechnological and clinical improvements. The generation of sequence data greatly outpaces the rate at which this data can be processed, analyzed, and understood. To address this discrepancy, he decided to launch a bioinformatics firm whose purpose is to assist in the processing, analysis, and interpretation of biological sequence data.

Shiva Sadeghpour: Junior Bioinformatician
Specializing in pipeline automation, Nextflow, and AWSShiva earned her bachelor's degree in Biology from UC Irvine and began her research journey at a neuroscience lab at Caltech, where she gained hands-on research experience in a large-scale lab environment. Discovering the power of computational biology, she pursued a master’s degree in Bioinformatics at Cal State LA, focusing on leveraging big data to study microbial communities and their complex interactions. Shiva is passionate about using bioinformatics tools and contributing to the development of new tools to uncover patterns in biological systems.
Dr. Ashley Cohen: Bioinformatics consultant
Specializing in microbial ecology, geochemistry, and PythonAshley earned her Masters degree in Geosciences and PhD in Marine Sciences with a concentration in microbiology at Stony Brook University in NY. Her background includes organic biogeochemistry, microbiology, ecology, and biostatistics. A combination of expertise in wet-lab and computational techniques makes Ashley an invaluable resource when it comes to generating, processing, and interpreting the many different forms biological data. Ashley is passionate about creating tools to solve common problems in microbiology workflows, tools that are accessible to users with no coding experience. Ashley has also enjoyed many years of outreach by advising undergraduate students in the laboratory and creating open-source python tutorials as part of the Bioinformatics Virtual Coordination Network.

Common Bioinformatics services
| Service | Description |
|---|---|
| Prokaryotic genome assembly and annotation | Reference-guided or de novo assembly using Unicycler, followed by a comprehensive annotation pipeline that includes de novo prediction of coding (CDS) and non-coding (tRNA, rRNA, tmRNA, miRNA) gene sequences by BAKTA. Predicted genes are then compared against a variety of databases, including KEGG, COG, CAZy, [Pfam/InterPro(https://www.ebi.ac.uk/interpro/), TIGRFAMs, and ISFinder. |
| Eukaryotic genome assembly and annotation | Reference-guided or de novo assembly using SPAdes and (if long-reads are available) Longstitch. If long reads are available, Flye is also used to build an initial draft assembly. If short-reads are available, polishing is then carried out using Pilon. Gene prediction is carried out using Braker and functional annotation using eggNOG-mapper. |
| Metagenome assembly | Optimized de novo assembly using metaSPAdes and Megahit. Other techniques can be considered depending on the data used. |
| Metagenome binning | Multiparametric binning using MetaBAT and DASTool, followed by bin evaulation with CheckM, SprayNPray, and Binarena. Other techniques can be considered depending on the data used. |
| Phylogenomics | Identification of single-copy genes using GToTree, and generation of a phylogenomic tree in the context of 100 most-closely related genomes available from NCBI's RefSeq database. |
| RNA seq and differential expression | Read mapping to reference genome using Bowtie2. Data is summarized into count tables using HTSeq, and differential expression analysis performed in DESeq2. Transcripts are reconstructed using Trinity. |
| Amplicon (16S) analysis | Analysis using Qiime2 and Dada2, producing an ASV/OTU table, sequences for each ASV/OTU in FASTA format, and taxonomic assignment for each sequence. |
| Amplicon Biostatistics and visualizations | Diversity calculations include alpha diversity indices (Shannon’s index, Pielou eveness) and beta diversity matrices (Jaccard, Bray-Curtis, Unifrac) derived from an appropriately rarefied OTU table and a binned metadata table. Beta diversity matrices are further processed by principal coordinate analysis, and those results are visualized with the metadata. Statistical testing against alpha diversity (Kruskall-Wallace tests) and beta diversity (PERMANOVA, ANOSIM) is also available. Rarified OTU tables can be transformed into relative abundances with the option of pseudo-counting and further clr-transformation, as well as ANCOM testing against binned metadata. |
| Taxonomic classification | Taxonomic profiling at the read level, using Kraken, and at the contig level (following a Unicycler assembly) using SprayNPray. This analysis also includes identification of the most closely related sequenced genomes from RefSeq/GenBank using Mash and construction of a phylogenomic tree using GToTree. |
| Virus identification | Identification of virus and phage sequences using VirSorter. |
| Consultation | MAB is available to provide advice, feedback, and assistance with pipeline development, software development, bioinformatics analyses, and data interpretation. A retainer for an extended period of time (e.g. weeks, months, and years) is possible at a discounted rate. Initial consultation session is free. |
| Letters of support and grant assistance | We are happy to assist/consult in grant preparation, particularly regarding any proposed bioinformatics. Additionally, MAB will, upon request, provide a letter of support in regard to any bioinformatics analysis or training that is included as part of the grant proposal. |
SEQUENCING AND BIOINFORMATICS
We partner with world-class sequencing centers to offer short-read (Illumina) sequencing and long-read (Nanopore) sequencing with PromethION.
COST BREAKDOWN
| Sample Type | Cost/Sample |
|---|---|
| bacterial genome | $120-150 |
| yeast genome | $190-245 |
| metagenomes and larger eukaryotic genomes | $300-800 |
| bacterial genome (hybrid-sequenced) | $300-550 |
| RNAseq (with rRNA depletion using RiboZero) | $309-455 |
Tell us about your data analysis needs below
| Common Service Offerings | Analyses Included |
|---|---|
| WHAT'S IN THE BAG | -strain-typing and serotyping -multi-locus sequence typing |
| RESISTANCE IS FUTILE | -variant-calling -antimicrobial compound and resistance discovery |
| DUDE WHERE'S MY TRANSPOSON | -Tn-Seq and genetic construct validation |
| AMPLICON/MICROBIOME | -16S/18S/ITS diversity analyses |
| GENOME ASSEMBLY/ANNOTATION | -prokaryotic genome assembly -de novo gene prediction and functional inference |
| RNA-SEQ | -differential expression analysis -transcriptome assembly -rRNA filtration. |
TRAINING AND WORKSHOPS
We are available to train biologists at all levels, from undergrad to professor, in bioinformatics techniques that are of interest to each respective lab. The scope and flexibility of training includes private lessons with individual researchers, as well as department-wide workshops. These can be virtual, in-person, or a combination of both.Please contact us for a customized lesson plan and quote.
| Tier | Description |
|---|---|
| Tutoring / Mentoring (One-on-One Support) | Personalized bioinformatics guidance for students or individual researchers. Ideal for learning new tools, troubleshooting analyses, or building confidence with coding and data workflows. Sessions can be structured as recurring mentoring, project-specific support, or skills-focused lessons. |
| Small Group Workshops (2–8 Participants) | Training sessions tailored for lab groups, research teams, or small cohorts. May include interactive coding demonstrations, workflow walk-throughs, and collaborative problem-solving exercises. Can be adapted to varying skill levels and research interests within the group. |
| Large Group Seminars or Course Modules (Department-Wide / Classroom) | Lecture-style presentations or structured instructional modules designed for university courses, seminar series, or institutional training events. Suitable for introducing foundational concepts, emerging methods, or domain-specific research applications to a broad audience. |
CONSULTING
At MAB, we specialize in providing expert consultation for large-scale academic and industry projects in environmental science. Our experienced team of scientific consultants bring a wealth of knowledge across various disciplines relevant to environmental science and microbial ecology. Whether you’re navigating complex regulatory landscapes, designing sustainable solutions, or advancing cutting-edge research, we deliver the expertise you need to achieve your goals. Our approach is collaborative and data-driven, helping you meet critical deadlines and objectives with confidence.
We provide a wide range of consulting services tailored to academic and industry needs (with rates ranging from $75-200/hr)
| Scope | Description |
|---|---|
| Project Structuring and Management | Comprehensive support for planning, executing, and managing large-scale environmental projects. |
| Scientific Expertise | Expert staff available to guide research design, data analysis, and interpretation for environmental studies. |
| Data Analysis and Modeling | Advanced computational and statistical analyses to predict environmental changes and outcomes. |
| Sustainability Solutions | Development of strategies for achieving long-term environmental and economic sustainability. |
SEMINARS
We also offer customized scientific seminars for academic departments, research institutes, professional societies, and industry teams seeking to deepen their understanding of cutting-edge topics in bioinformatics, microbiology, and environmental genomics. Seminars can range from high-level conceptual overviews to data-rich, research-focused presentations tailored to your audience’s background and interests. Formats include invited talks, guest lectures for university courses, seminar-series presentations, or small-group discussion sessions. Whether you are looking to introduce new analytical approaches, highlight emerging discoveries, or spark interdisciplinary collaboration, we can design and deliver a seminar that aligns with your goals and engages your community.
| Seminar type | Description |
|---|---|
| Foundations of Bioinformatics | An accessible introduction to core computational methods used in modern biological research. Suitable for undergraduate students, research labs beginning to adopt computational workflows, and interdisciplinary audiences.. |
| Genomics and Environmental Microbiology Seminar | A research-focused presentation examining microbial communities, symbiosis, metagenomics, and environmental genomics, tailored for academic seminar series and departmental talks. |
| Applied Metagenomics for Research and Industry | A seminar emphasizing practical applications of metagenomics, including data interpretation, troubleshooting, and workflow design for environmental, clinical, and industrial use cases. |
| Emerging Tools and Methods in Molecular Ecology | A forward-looking seminar exploring recent advances in sequencing technologies, computational tools, and multi-omics integration, with discussion of future trends and research opportunities. |
CONTACT US FOR A PERSONALIZED QUOTE
INTERESTED IN WORKING WITH US?
NCBI DATA SUBMISSION
| Sample Type | Repository |
|---|---|
| Genome | GenBank and RefSeq |
| FASTQ reads | Sequence Read Archive (SRA) |
| Metadata | FigShare, GitHub, etc. |
We know how tedious and complicated data submission to NCBI can be. But reproducibility and open access of data is essential to many ongoing projects that rely on and add upon previous work. To this end, we offer, as a service, submission of samples to NCBI and other public repositories (e.g. GitHub, FigShare).
PROBE DESIGN FOR RNA-FISH
Fluorescence in situ hybridization (FISH) is a powerful technique that allows for simultaneous visualization, phylogenetic identification, and enumeration of individual microbial cells. FISH is therefore often a critical step in many cutting-edge microbiology workflows that separate cells of a particular phylogenetic affiliation for single-cell amplicon or genome sequencing or that localize and interrogate those cells using microspectroscopy methods such as NanoSIMS (determine natural stable isotopic relative abundances) or Raman (determine which cells have taken up an isotopically spiked substrate).This method entails irreversibly binding a fluorescently labelled probe to permeabilized cells’ ribosomal RNA. Probes are 16-23 mers with a base sequence that is complimentary to a consensus region- a sequence that is conserved among and unique to a phylogenetic target group- within a target phylogenetic group’s 16S or 23S gene. Probes will typically hybridize against sequences with a 0 or 1 base pair mismatch, so that many must be combined with competitor probes- non-fluorescent probes that bind to non-target sequences with a one-base mismatch so that they are “unavailable” to the FISH probe. Some phylogenetic groups also require probe “cocktails” for near-total coverage (for example, Deltaproteobacteria). For robust experimental results, it is essential that the FISH probes have a high specificity (unique to target phylogenetic group) and coverage (accounts for a high percentage of target group sequences) against a curated database of high-quality 16S or 23S sequences such as SILVA or GreenGenes, and against the user’s sample 16S or 23S sequences. While there are online tools that aid in these analyses to a degree, such as TestProbe, they lack several important services. These include calculating competitor probe-adjusted coverages and specificities, coverages and specificities of “cocktails” and testing the probe(s) and competitor(s) against the user’s sample library.Our service accomplishes all of this in an easily executable python pipeline and has a variety of optional outputs. These include database target and non-target accessions and sequences, the position(s) and base(s) of the most common mismatches, suggested additional competitor probes, and probe testing against the user’s amplicon libraries through alignments with high-quality reference sequences and consensus region checks.
SAMPLE PLOTS
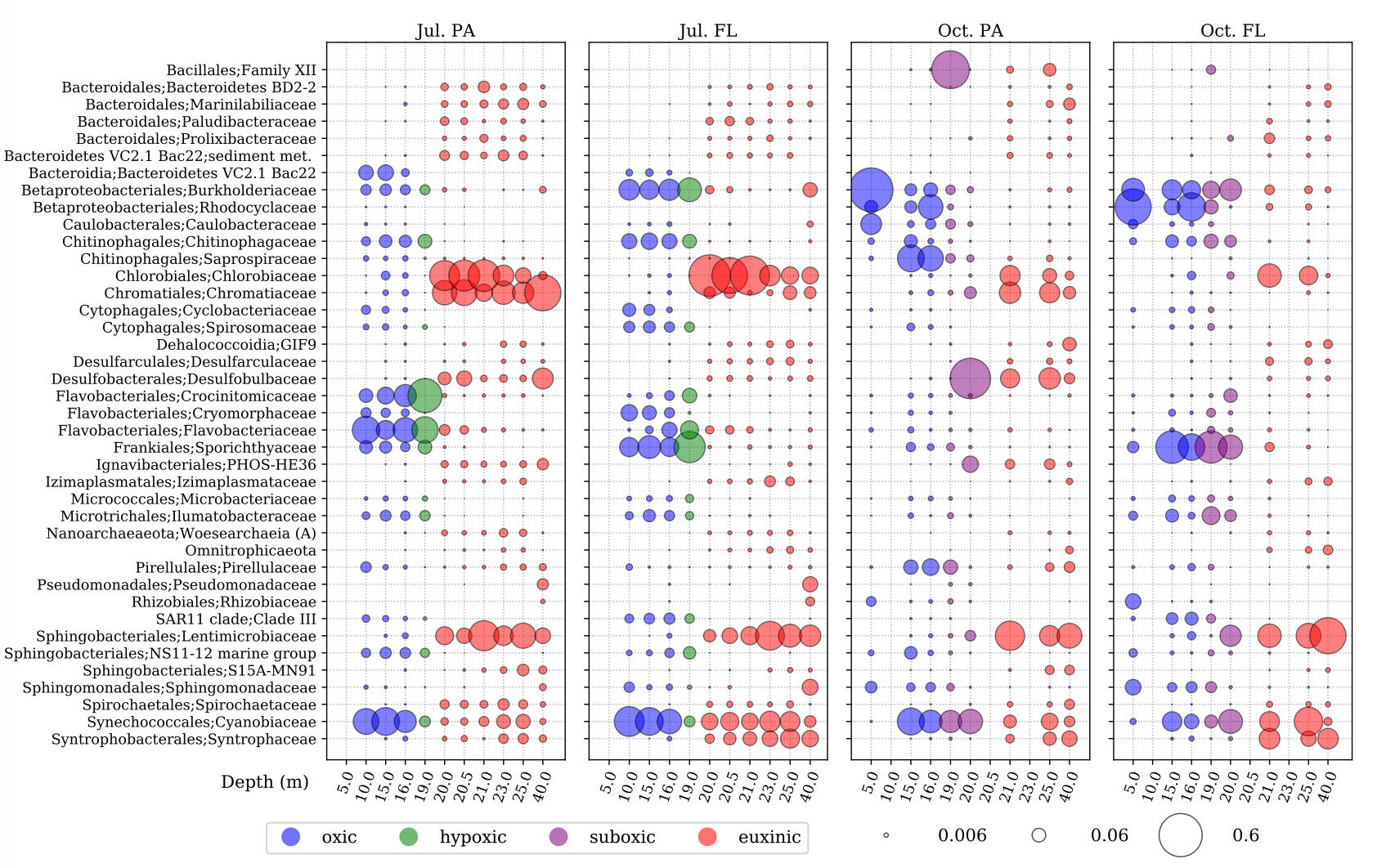
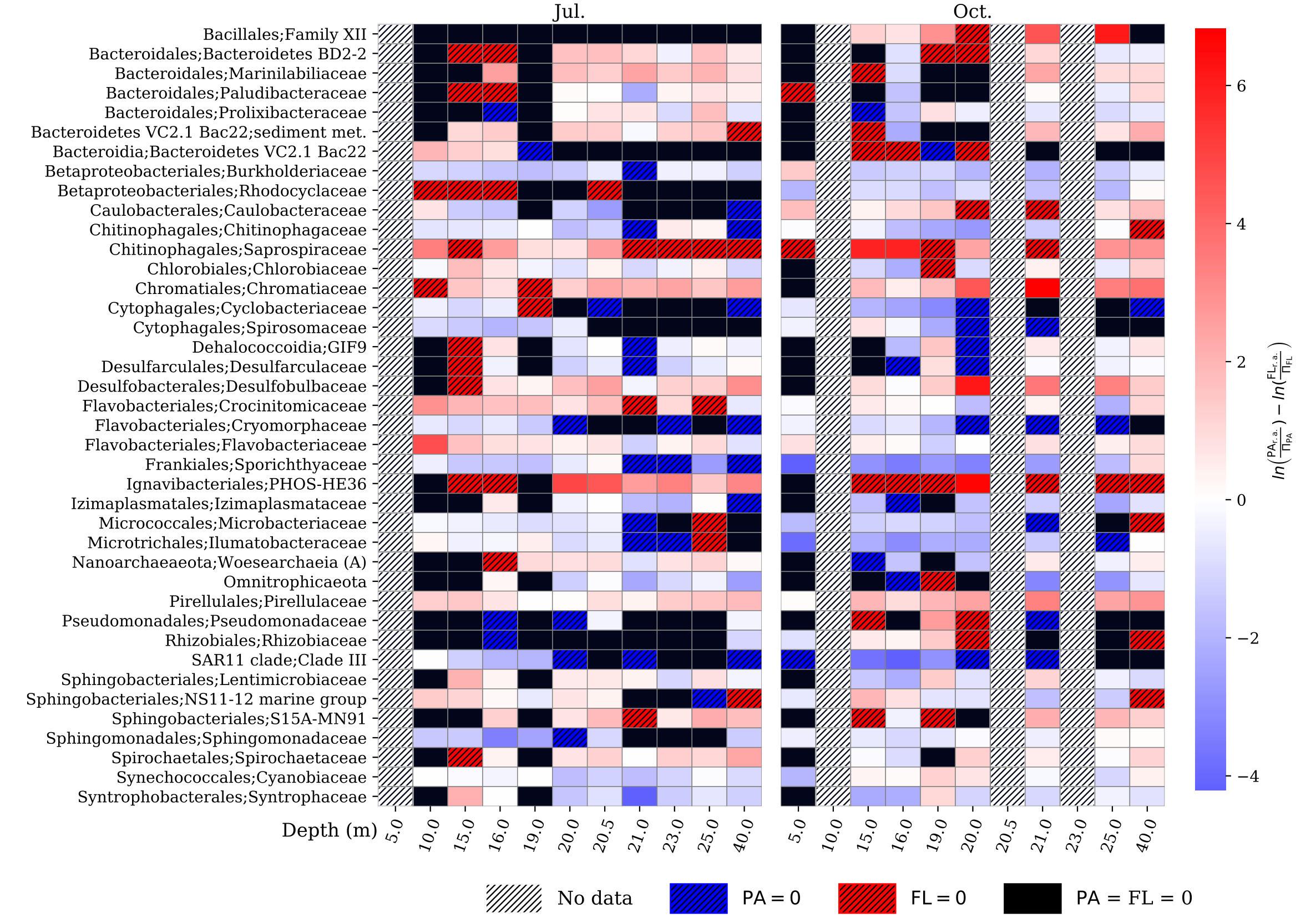
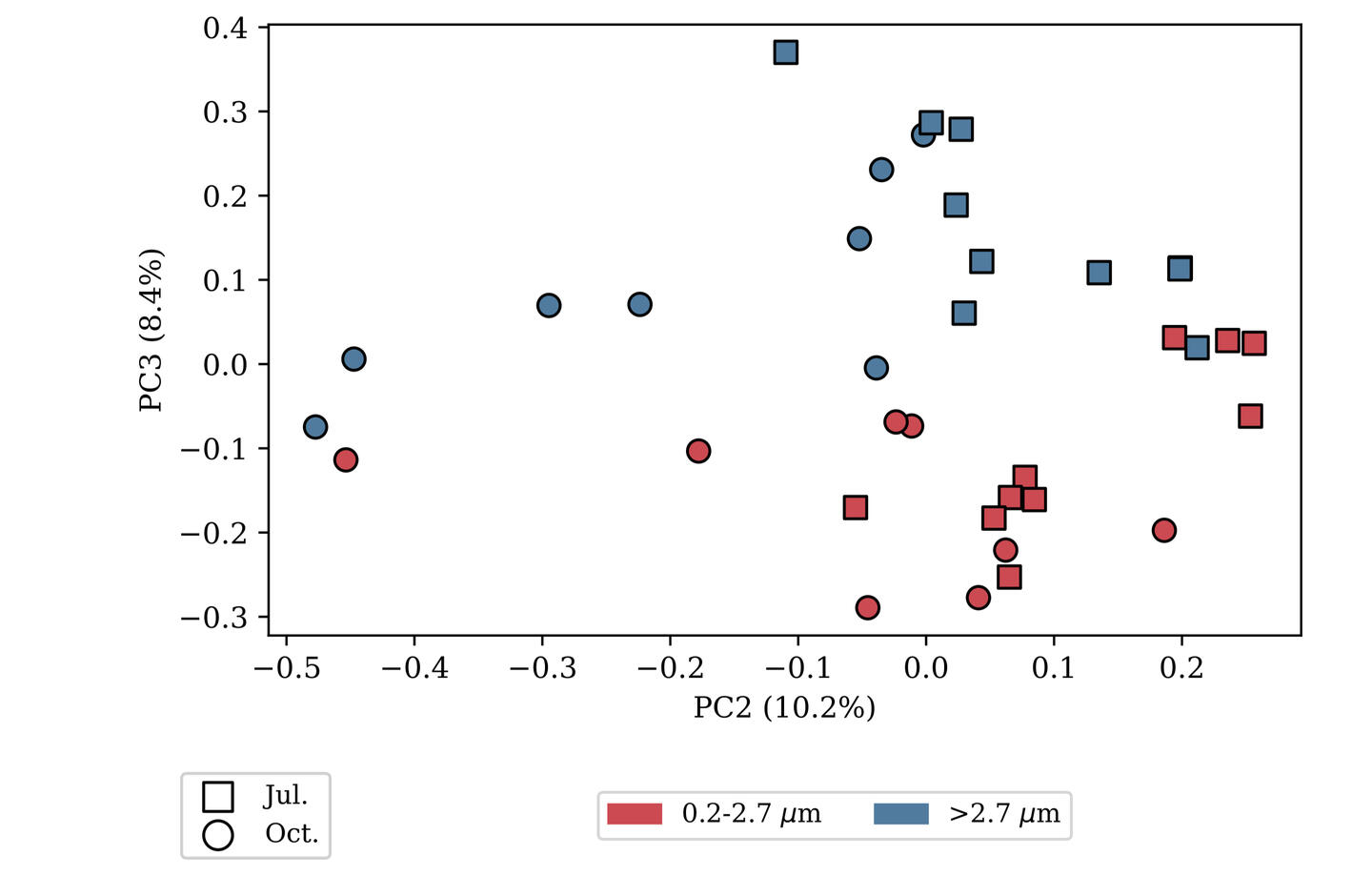
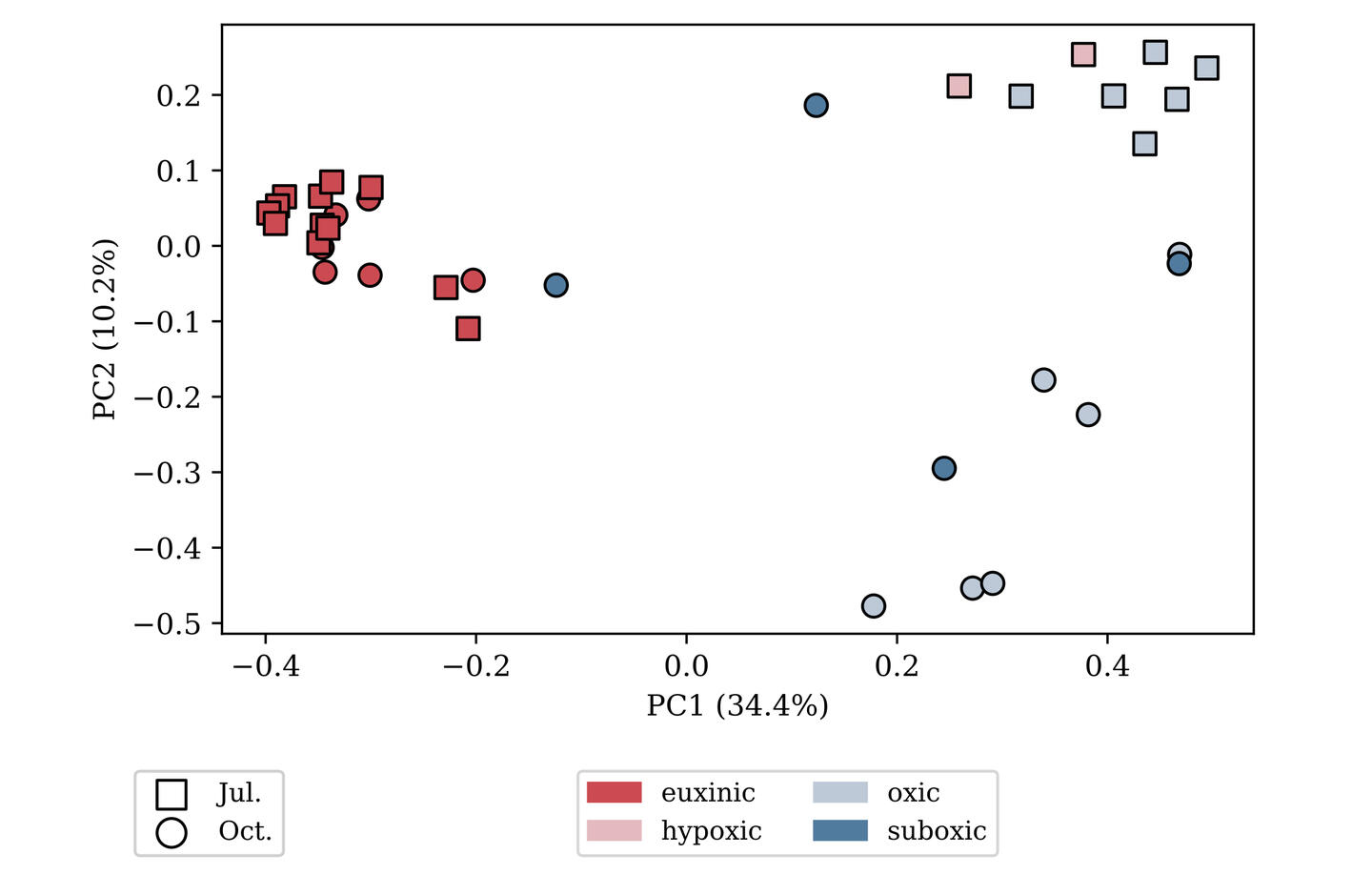
(bioinformatics as a service)
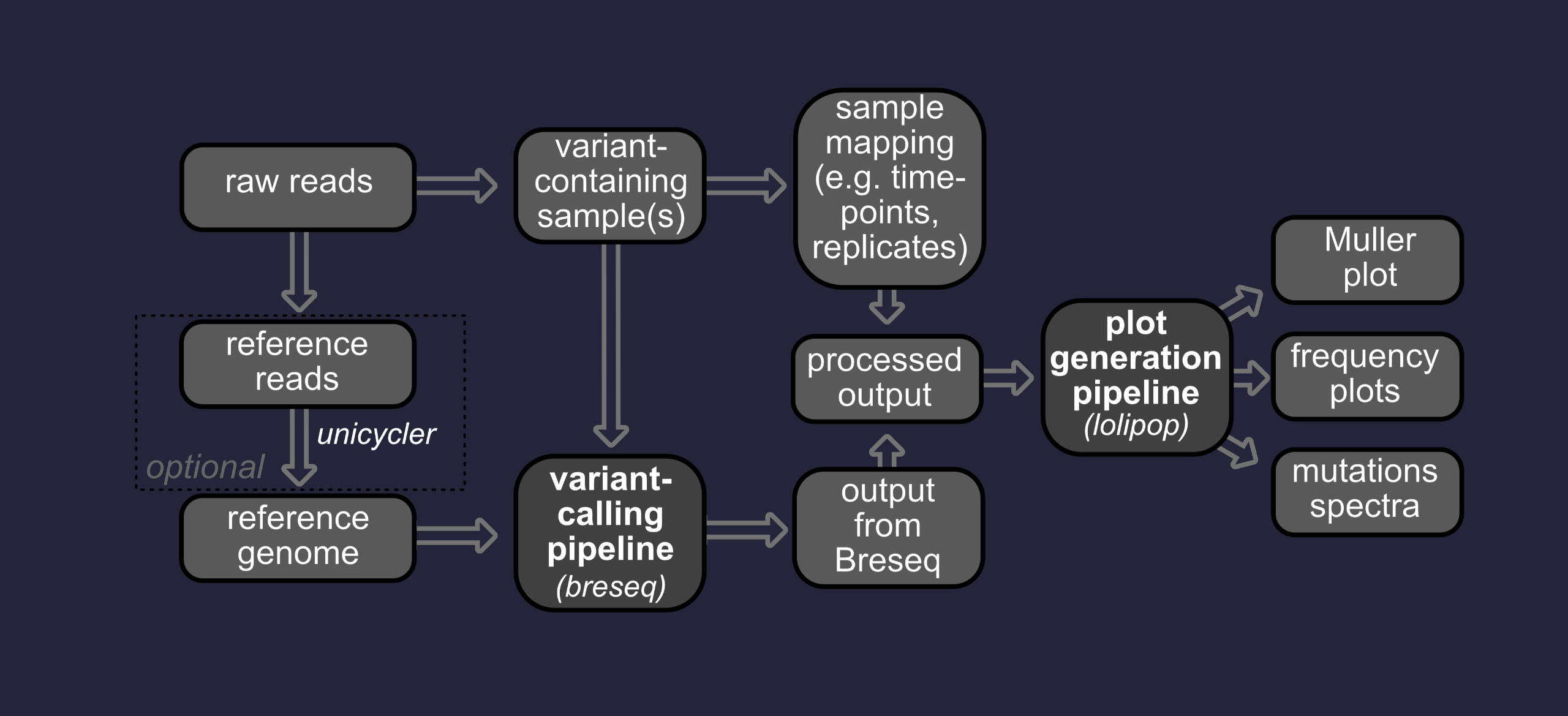
variant calling
Variant calling is the process of identifying mutations in an evolved lineage. Mutations are predicted by identifying changes in the genome sequence over the course of an experiment (e.g. evolve-and-resequence)
Please contact us for a customized lesson plan and quote.
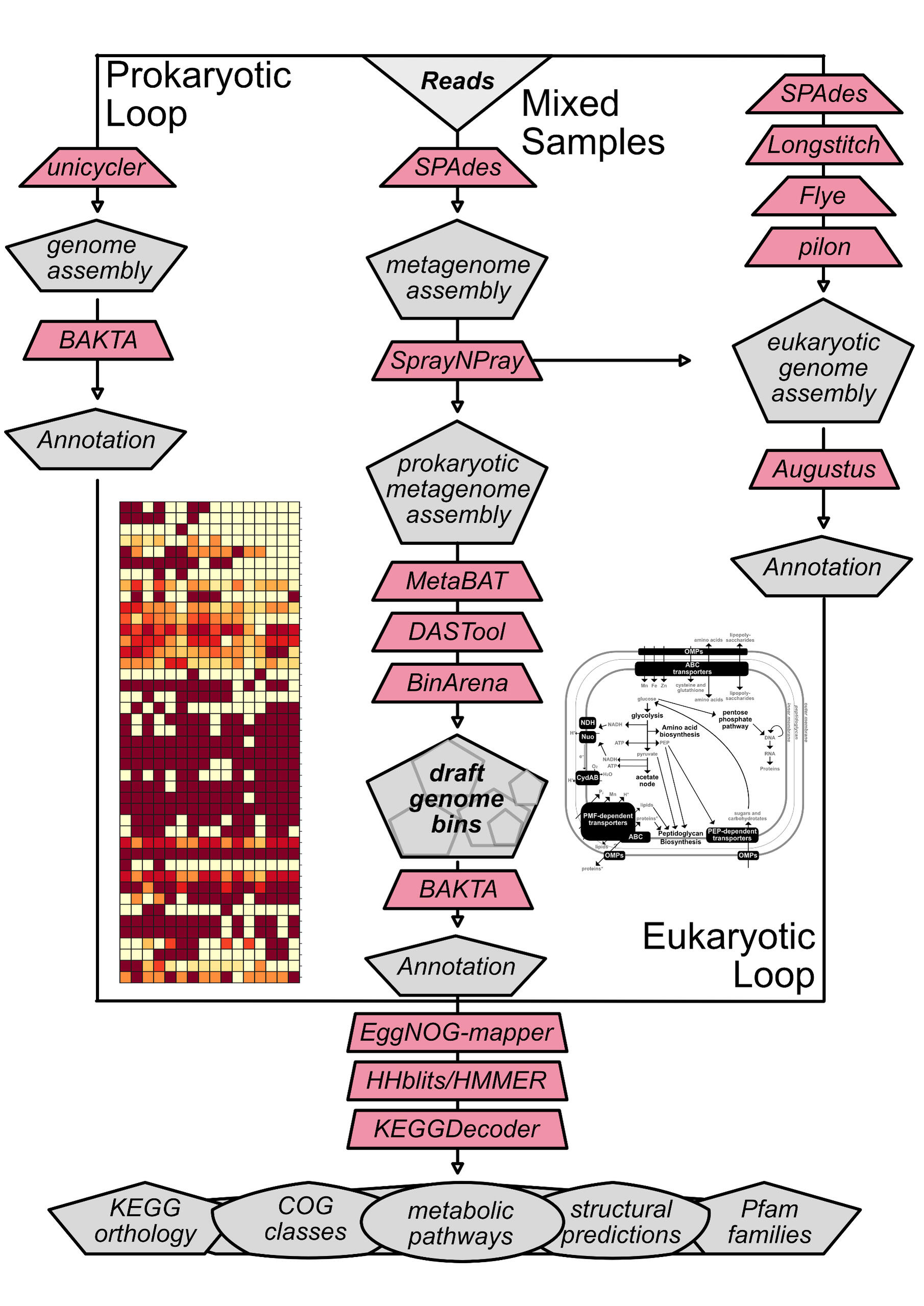
(meta)genomics
Genomes are routinely sequenced with methods that involve shearing DNA into smaller fragments prior to sequencing. Computational pipelines exist that assembly these reads back together based on overlapping sequences.
prokaryotic genome assembly
Reference-guided or de novo assembly using Unicycler, followed by a comprehensive annotation pipeline that includes de novo prediction of coding (CDS) and non-coding (tRNA, rRNA, tmRNA, miRNA) gene sequences by BAKTA. Predicted genes are then compared against a variety of databases, including KEGG, COG, CAZy, [Pfam/InterPro(https://www.ebi.ac.uk/interpro/), TIGRFAMs, and ISFinder.
eukaryotic genome assembly
Reference-guided or de novo assembly using SPAdes and (if long-reads are available) Longstitch. If long reads are available, Flye is also used to build an initial draft assembly. If short-reads are available, polishing is then carried out using Pilon. Gene prediction is carried out using Braker and functional annotation using eggNOG-mapper.
Metagenomics
Optimized de novo assembly using metaSPAdes and Megahit. Other techniques can be considered depending on the data used.Multiparametric binning using MetaBAT and DASTool, followed by bin evaulation with CheckM, SprayNPray, and Binarena. Other techniques can be considered depending on the data used.Identification of single-copy genes using GToTree, and generation of a phylogenomic tree in the context of 100 most-closely related genomes available from NCBI's RefSeq database.
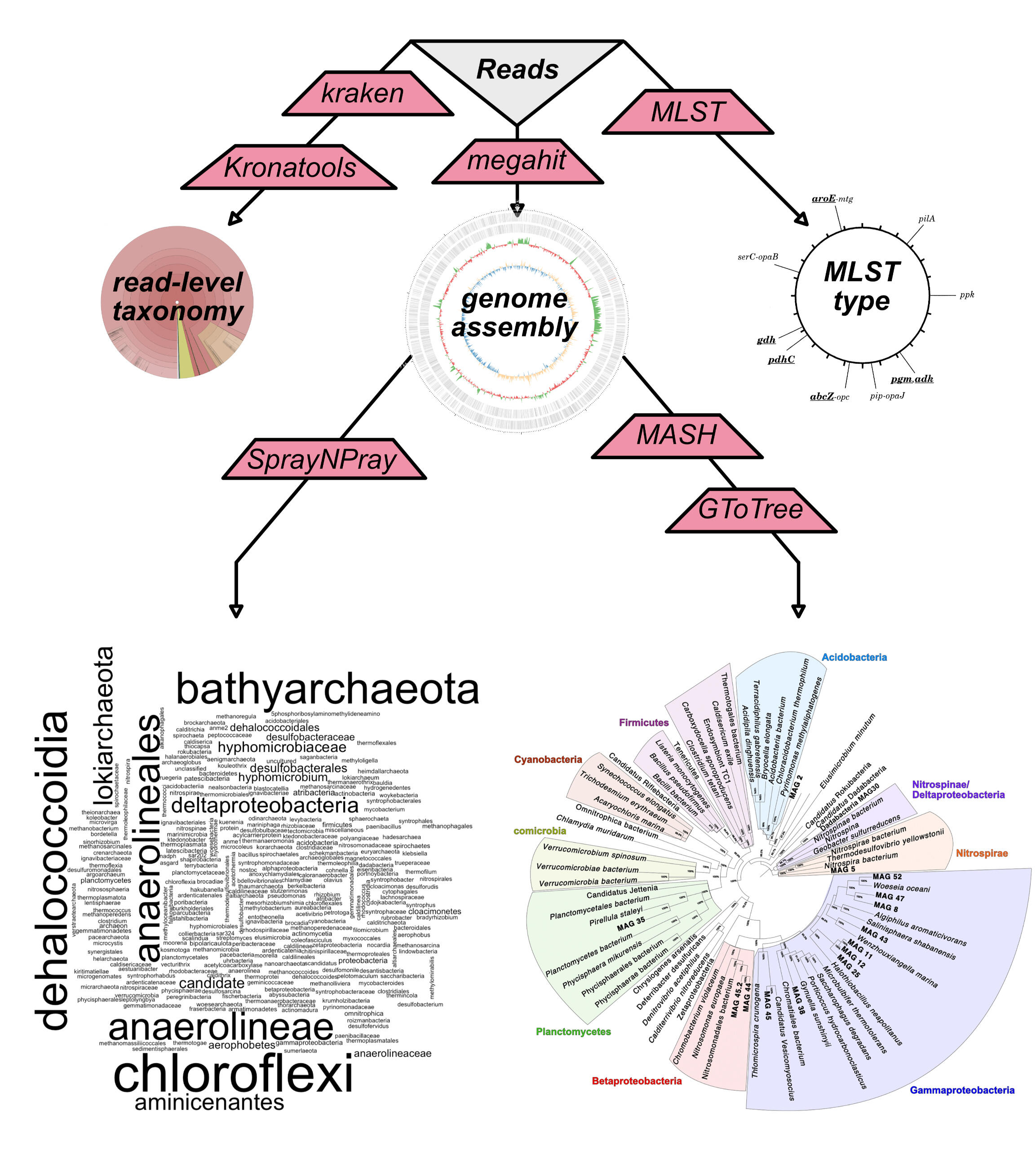
phylogenomics
Taxonomic profiling at the read level, using Kraken, and at the contig level (following a Unicycler assembly) using SprayNPray. This analysis also includes identification of the most closely related sequenced genomes from RefSeq/GenBank using Mash and construction of a phylogenomic tree using GToTree.
microbiome profiling
Analysis using Qiime2 and Dada2, producing an ASV/OTU table, sequences for each ASV/OTU in FASTA format, and taxonomic assignment for each sequence.Diversity calculations include alpha diversity indices (Shannon’s index, Pielou eveness) and beta diversity matrices (Jaccard, Bray-Curtis, Unifrac) derived from an appropriately rarefied OTU table and a binned metadata table. Beta diversity matrices are further processed by principal coordinate analysis, and those results are visualized with the metadata. Statistical testing against alpha diversity (Kruskall-Wallace tests) and beta diversity (PERMANOVA, ANOSIM) is also available. Rarified OTU tables can be transformed into relative abundances with the option of pseudo-counting and further clr-transformation, as well as ANCOM testing against binned metadata.
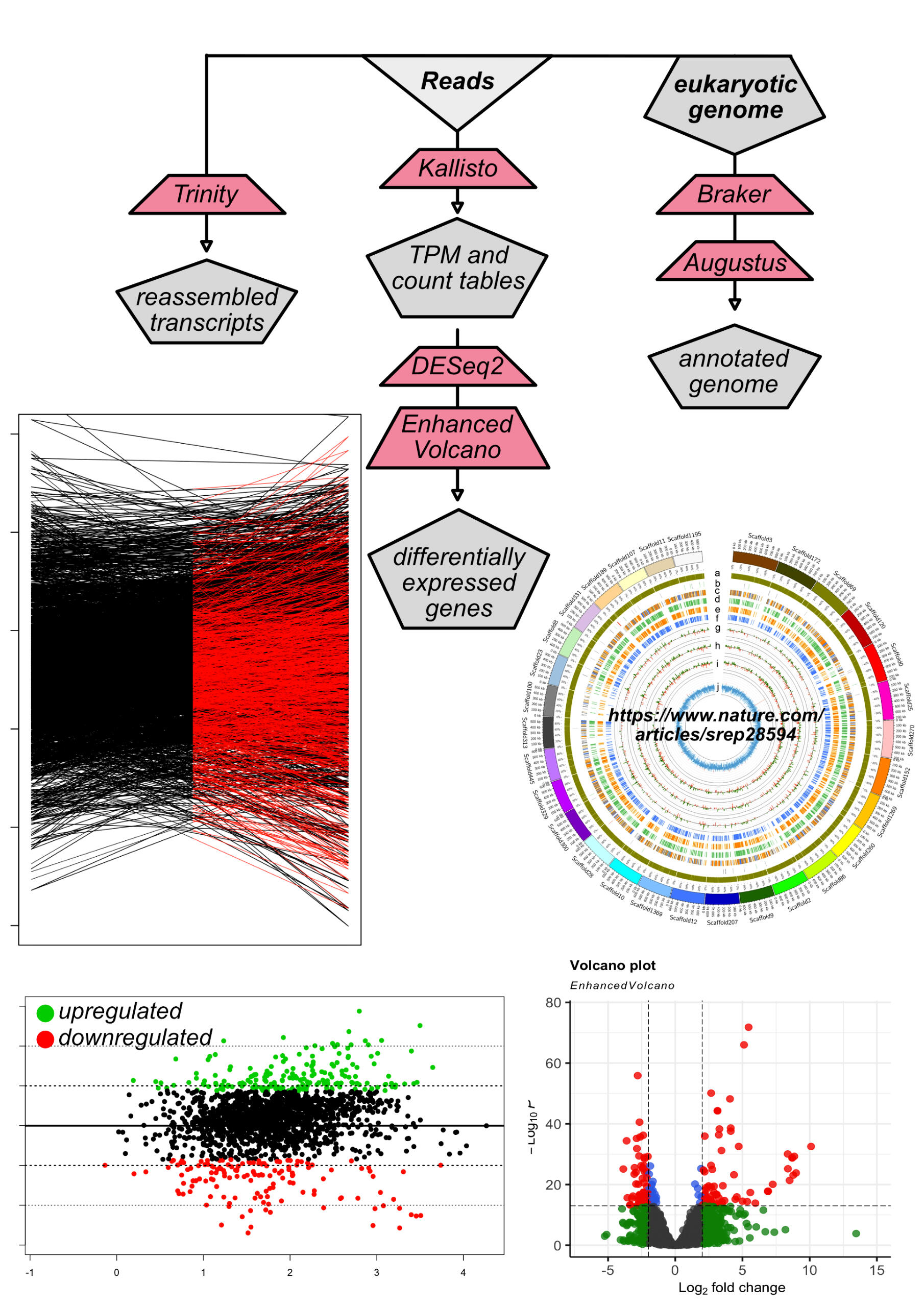
transcriptomics
Read mapping to reference genome using Bowtie2. Data is summarized into count tables using HTSeq, and differential expression analysis performed in DESeq2. Transcripts are reconstructed using Trinity.
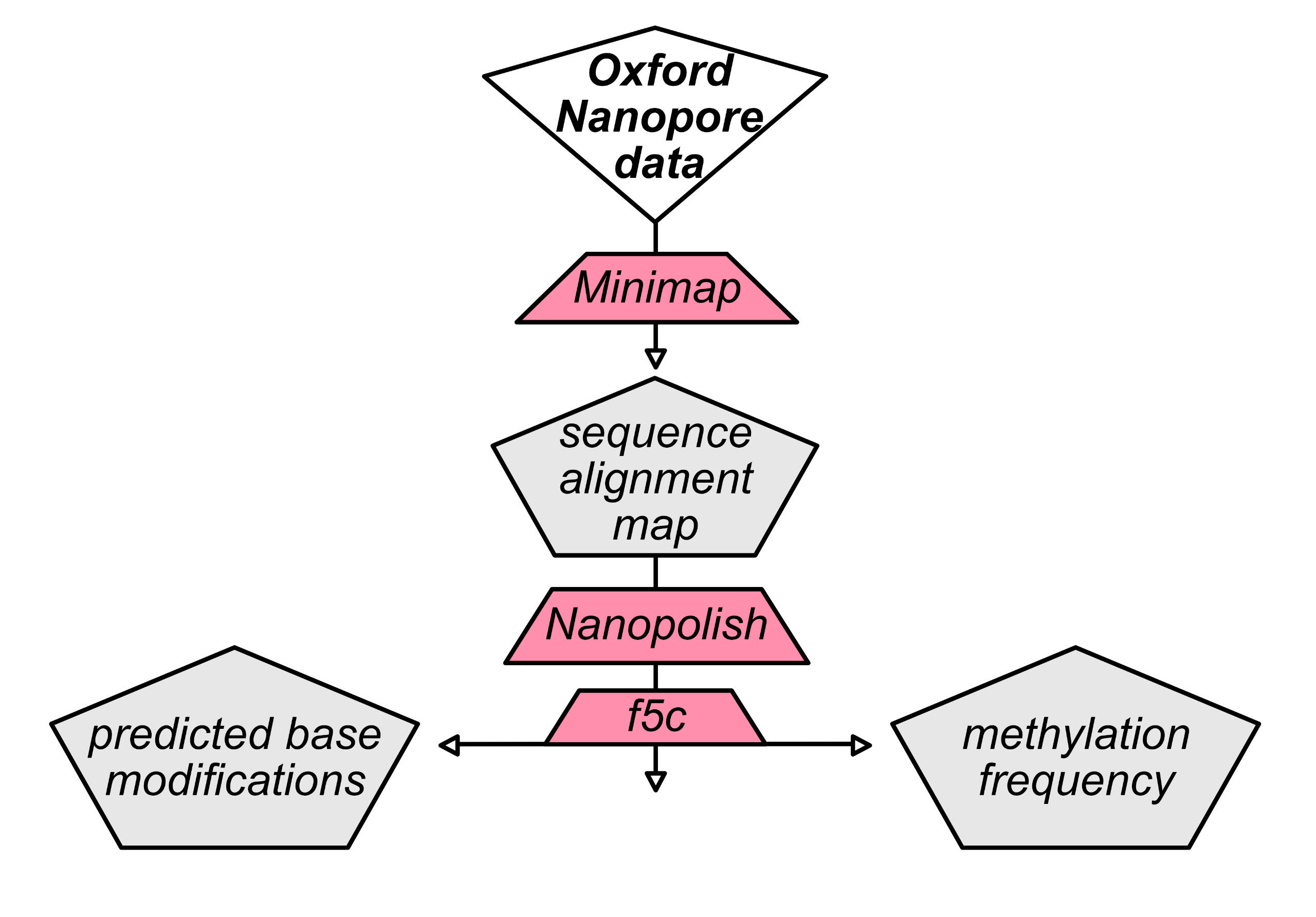
methylation prediction
Text
Our Privacy Policy
We respect your privacy and are committed to protecting your personal data. This Privacy Policy outlines how we collect, use, and safeguard your personal information when you use our website (midauthorbio.com) and our associated web applications, like omix.midauthorbio.com and magiclamp.midauthorbio.comWe collect only the following personal information:1) Name2) Email3) Genomic data provided explicitly by users for analysisHow Information is Collected and Used
1) Personal information is collected directly from you via submission forms on our website.2) We use the information you provide solely to perform bioinformatics analysis on your submitted genomic data.3) Your personal information is not used for marketing purposes unless you explicitly opt into our subscriber list, in which case you may receive occasional emails from us.4) Your information or data will never be used internally in any capacity, including in AI-related applications.Data Storage and Security
Your data is securely transferred to and stored on Amazon Web Services (AWS). We take the following measures to protect your data:1) Secure transfer protocols (HTTPS/SSL)2) Encryption during data storage and transmission3) Strictly controlled access via restricted IAM roles4) Automated weekly deletion of all user-submitted genomic data5) Active monitoring through AWS CloudTrail and AWS CloudWatchSharing of Your Information
We do not share your personal or genomic data with any third parties except where necessary to perform our services (AWS for storage and processing). Your information is never sold or shared for marketing purposes.User Access and Rights
If you wish to access, correct, or delete any of your personal data, you can reach out to us via our contact form at https://midauthorbio.com or by email at [email protected].Cookies and Tracking
We do not use cookies or any other tracking technologies on our website.Compliance with Laws
Currently, we are not subject to specific privacy laws such as GDPR or HIPAA but maintain standards that respect user privacy and data security.Changes to This Policy
We may update this Privacy Policy from time to time. Any changes will be posted directly to our website and communicated via email to subscribers if applicable.Contact Us
If you have any questions or concerns about this Privacy Policy or our data practices, please contact us at:Middle Author Bioinformatics
Pittsburgh, PA, USA
Email: [email protected]